


















































 Islands")


















































")






")
















































")
























































































")
")





 Germany
Germany
 Japan
Japan
 United Kingdom
United Kingdom
 China
China
CEP104 (NM_014704) Human Tagged ORF Clone
CAT#: RC212710L4
- LentiORF®
Lenti ORF clone of Human centrosomal protein 104kDa (CEP104), mGFP tagged
"NM_014704" in other vectors (4)
Product Images
Specifications
| Product Data | |
| Type | Human Tagged ORF Clone |
| Tag | mGFP |
| Symbol | CEP104 |
| Synonyms | CFAP256; GlyBP; JBTS25; KIAA0562; ROC22 |
| Vector | pLenti-C-mGFP-P2A-Puro |
| E. coli Selection | Chloramphenicol (34 ug/mL) |
| Mammalian Cell Selection | Puromycin |
| Sequence Data |
The ORF insert of this clone is exactly the same as(RC212710).
|
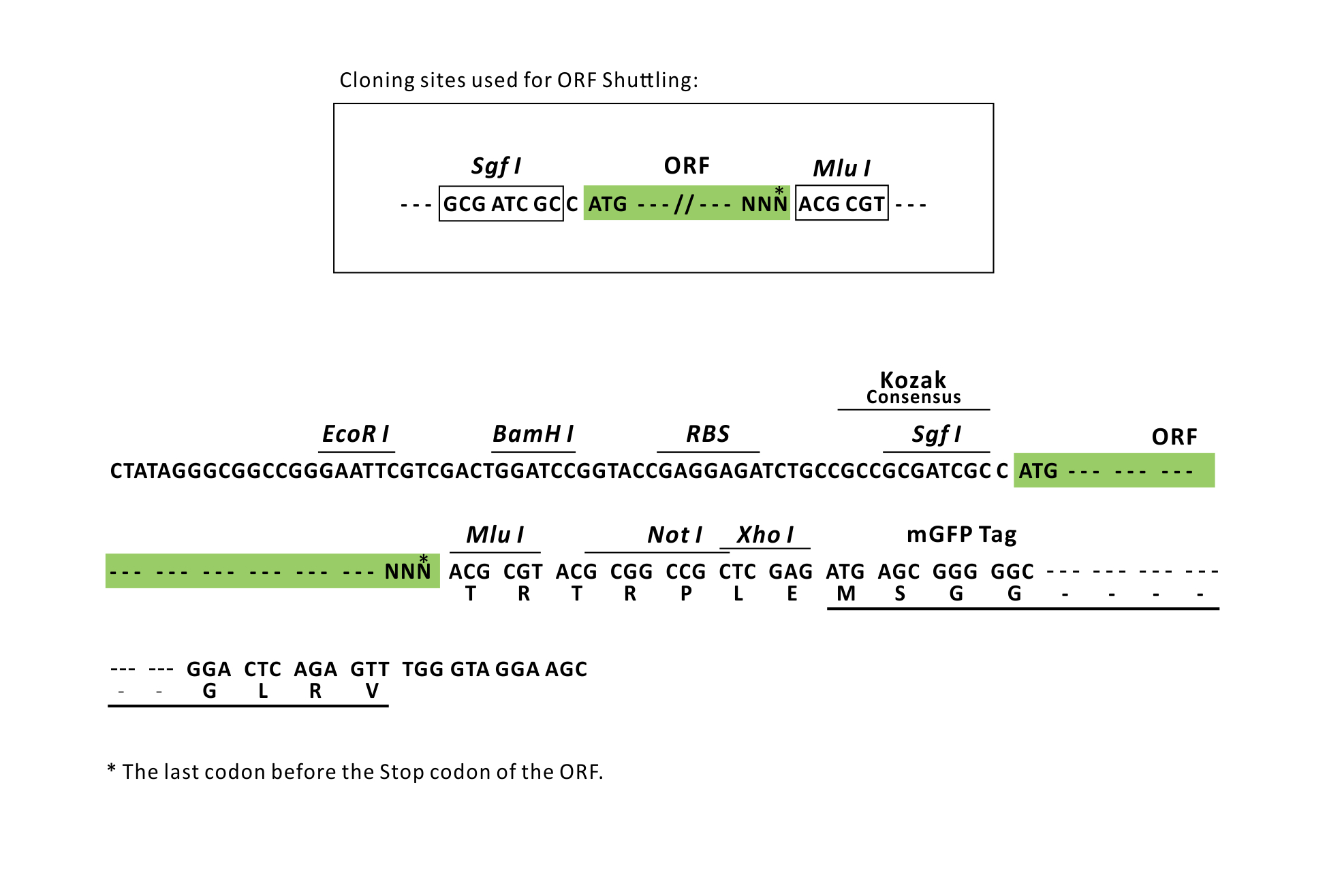
| Restriction Sites | SgfI-MluI Cloning Scheme for this gene |
| ACCN | NM_014704 |
| ORF Size | 2775 bp |
| OTI Disclaimer | The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info |
| OTI Annotation | This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. |
| Reference Data | |
| RefSeq | NM_014704.1, NP_055519.1 |
| RefSeq Size | 5848 |
| RefSeq ORF | 2778 |
| Locus ID | 9731 |
| MW | 104.3 kDa |
| Gene Summary | This gene encodes a centrosomal protein required for ciliogenesis and for ciliary tip structural integrity. The mammalian protein contains three amino-terminal hydrophobic domains, two glycosylation sites, four cysteine-rich motifs, and two regions with homology to the glutamate receptor ionotropic, NMDA 1 protein. During ciliogenesis, the encoded protein translocates from the distal tips of the centrioles to the tip of the elongating cilium. Knockdown of the protein in human retinal pigment cells results in severe defects in ciliogenesis with structural deformities at the ciliary tips. Allelic variants of this gene are associated with the autosomal-recessive disorder Joubert syndrome, which is characterized by a distinctive mid-hindbrain and cerebellar malformation, oculomotor apraxia, irregular breathing, developmental delay, and ataxia. [provided by RefSeq, Feb 2016] |
{kind=link}
Documents
| Product Manuals |
| FAQs |
| SDS |
Resources
Other Versions
| SKU | Description | Size | Price |
|---|---|---|---|
| SC114917 | CEP104 (untagged)-Human centrosomal protein 104kDa (CEP104) |
USD 1,880.00 |
|
| RC212710 | CEP104 (Myc-DDK-tagged)-Human centrosomal protein 104kDa (CEP104) |
USD 1,030.00 |
|
| RG212710 | CEP104 (GFP-tagged) - Human centrosomal protein 104kDa (CEP104) |
USD 1,130.00 |
|
| RC212710L3 | Lenti ORF clone of Human centrosomal protein 104kDa (CEP104), Myc-DDK-tagged |
USD 1,230.00 |
{0} Product Review(s)
Be the first one to submit a review
